
Приложение 1 к GMP ЕС, редакция 2020 г. Производство стерильных лекарственных средств
09.03.2020by svs-artaЧистые помещения

Расширение со 127 пунктов в редакции 2008 года до 300 пунктов в редакции 2020 года, будет ли Приложение №1 менее двусмысленным?
Пересмотренное руководство требует, чтобы компании официально задокументировали стратегию контроля контаминации, в которой учитываются все технические и организационные меры, необходимые для обеспечения соответствия стерильных продуктов их критическим характеристикам качества. Этот обзорный документ предназначен для предоставления краткого обзора основных изменений и расширенных требований в удобном для чтения формате.
Введение

В фармацевтической промышленности производство стерильных лекарственных средств рассматривается большинством как операции с высоким риском из-за риска заражения продукта и последующего риска для пациента из-за введения продукта, который не подходит для предполагаемого использования. Риск того, что нестерильный продукт попадет к пациенту, является реальным риском, который привел к гибели пациентов во время нескольких нежелательных событий, включая инцидент в Давенпорте в 1971 году и Центр компаундирования Новой Англии в 2012 году. Это привело к сохранению потребности для регулирования производства стерильных лекарственных средств и необходимости в специальном нормативном руководстве для определения минимальных требований, которым должны следовать производители фармацевтической продукции, в форме Приложения 1 к основному руководству GMP.
В настоящее время Приложение 1 находится на пересмотре, и первый проект для консультаций был выпущен в 2017 году для общественного рассмотрения и комментариев. В состав ревизионного комитета, ответственного за последний проект, входят EMA, ВОЗ и PICS, а также вклад FDA в разработку согласованного руководства. С тех пор, как был выпущен первый проект, отрасль предоставила значительное количество комментариев, в результате чего в феврале 2020 года был выпущен второй проект, в который были включены комментарии из первоначального проекта. Затем следует дальнейший период общественных консультаций и окончательного рассмотрения комитетом перед выпуском окончательного документа. За последние 12 лет, начиная с версии 2008 года, были достигнуты значительные успехи в технологиях и изменениях в способах работы отрасли, в результате чего руководство GMP устарело, следовательно, возникла необходимость в его пересмотре.
Последний проект консультации – это переписывание
Задача регулирующего комитета, готовящего руководство, состоит в том, чтобы подготовить документ, который устанавливает минимальные требования для соответствия GMP, которым должен следовать каждый производитель, который намеревается продавать свою продукцию в Европу, или обосновывать альтернативный подход. Цель не в том, чтобы достичь идеального документа, а в достижении консенсуса по окончательному тексту перед выпуском. Текст должен быть достаточно открытым, чтобы учесть внедрение новых технологий и позволить производителям гибко применять альтернативные методы работы, при этом соблюдая фундаментальные принципы GMP, поэтому он не может быть чрезмерно предписывающим.
Проблема с рекомендациями GMP сводится к интерпретации текста и отдельных пунктов. Интерпретация пунктов, в свою очередь, сводится к опыту, культурным особенностям и перспективам человека или организации, читающим его, а также к тому, как воспринимаются изменения, влияющие на их производственные операции. Серьезные изменения в правилах или отсутствие ясности в интерпретации повышенных требований могут привести к необходимости модификаций оборудования или производственных операций или привести к недостаткам при следующей проверке GMP, поэтому это становится очень эмоциональной темой. Этот документ не предназначен для детального изучения формулировок, принятых в отдельных пунктах последнего проекта руководства.
Двумя наиболее значительными изменениями в документе являются требование к компаниям производить стерильные продукты с использованием принципов управления рисками для качества (QRM) и внедрять стратегию контроля загрязнения на всем предприятии.
Двумя наиболее значительными изменениями в документе являются требование к компаниям производить стерильные продукты с использованием принципов управления рисками для качества (QRM) и внедрять стратегию контроля загрязнения на предприятии. Это пример того, что нынешний текст Приложения 1 2008 г. устарел, поскольку использование принципов QRM при принятии решений в фармацевтическом производстве было нормативным требованием в течение нескольких лет. В Honeyman Group мы в течение нескольких лет проводим обучение по внедрению стратегии контроля загрязнения на предприятии с использованием инструментов оценки рисков, таких как HACCP, в нашем курсе «Критические факторы для производства стерильных продуктов». и мы видим, что многие компании выполнили эти оценки, поэтому правила обновляются, чтобы отразить текущую отраслевую практику. В новой редакции Приложения 1 теперь представлена структура стратегии контроля загрязнения (CCS) и примеры того, когда и где применять принципы управления рисками, и они внимательно изучаются промышленностью в течение периода консультаций.
Стратегия контроля загрязнения (CCS)
В пересмотренном Приложении определен принцип CCS. Рекомендуемый подход к CCS следует основным принципам HACCP, когда продукт и процесс анализируются целиком, чтобы понять работу каждой единицы и оценить потенциальные опасности загрязнения, а также на каждом этапе и меры по предотвращению возникновения опасности. Опасности четко определены в руководстве (микробиологические, эндотоксины и твердые частицы). Первоначальная сфера применения Приложения 1 касалась только стерильных продуктов, однако объем документа был расширен, в результате чего принципы, определенные в документе, могут быть применены к нестерильным продуктам. Таким образом, на самом деле те же принципы HACCP могут быть применены для оценки риска загрязнения продукта и валидации очистки при производстве нестерильных продуктов, хотя инструменты оценки риска должны быть изменены для этой цели. В пересмотренном Приложении излагаются элементы, которые следует учитывать в задокументированной стратегии CCS, которые включают комплекс технических мер, включая объект, конструкцию оборудования и материалы, в сочетании с рядом организационных мер, включая системы качества, квалификацию поставщиков и оборудования и мониторинг процесса.
Краткое изложение элементов CCS включено в Таблицу 1. Само собой разумеется, что здесь есть оговорка, что это основные элементы, которые следует учитывать в задокументированной CCS, но CCS не должна ограничиваться только этими элементами. Это подчеркивает открытый характер Приложения и то, что цель состоит в том, чтобы предоставить минимальный набор требований.
Таблица 1 Технические и организационные элементы CCS
| Технические меры | Организационные меры |
| Дизайн завода и процесса | Утверждение поставщика |
| Помещения и оборудование | Оценка рисков процесса |
| Утилиты | Проверка процесса |
| Контроль сырья | Профилактическое обслуживание |
| Контейнеры для продуктов и крышки | Системы качества
|
| Аутсорсинговые услуги | |
| Очистка и дезинфекция | |
| Системы мониторинга |
Фармацевтическая система качества (PQS) и оценка рисков
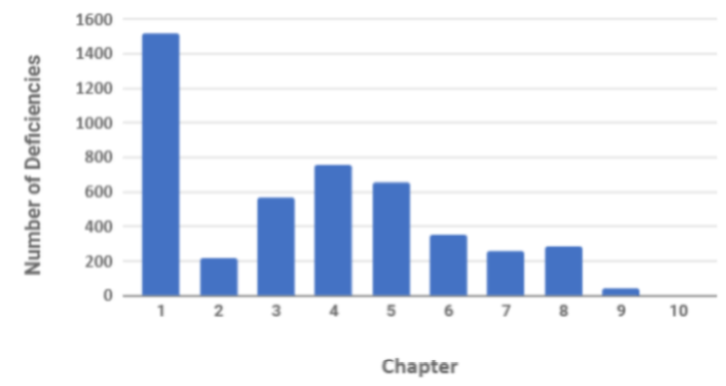
Что касается организационных мер, требования к системе качества были значительно пересмотрены в последней редакции с выделенным разделом о PQS. Исторически и в настоящее время наибольшее количество наблюдаемых недостатков инспекций связано с неадекватностью PQS и документации событий и расследований. При рассмотрении данных о недостатках инспекций MHRA 2018 это становится очевидным при наличии 1500 недостатков, связанных с несоблюдением главы 1.
График 1 Недостатки инспекции GMP по главам в 2018 г. ( исходный веб-сайт MHRA)
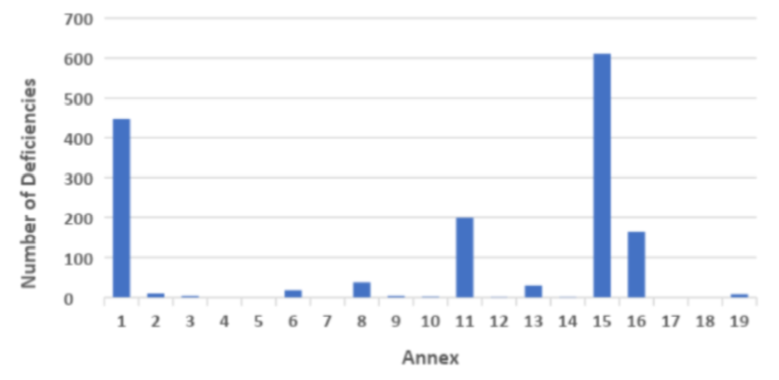
Что касается недостатков, относящихся к техническим аспектам, то наиболее часто наблюдались недостатки, связанные с стерильными продуктами Приложения 1, Валидацией Приложения 15 и выпуском КП по Приложению 16.
График 2 Недостатки инспекции GMP по приложениям в 2018 г. ( исходный веб-сайт MHRA)

В новом разделе PQS разъясняются ожидания в отношении эффективной системы качества, которая включает в себя обеспечение наличия у организации достаточных знаний и опыта в отношении продукции, которую она производит, и документирования решений. Это требование кажется очевидным, но компаниям может быть сложно удержать персонал из-за активности в отрасли и, конечно, когда персонал уходит, знания теряются, если процесс принятия решений не был задокументирован. Основная идея здесь состоит в том, чтобы понять продукт и процесс достаточно подробно, чтобы иметь возможность эффективно оценивать риски для качества продукта, связанные с загрязнением, и обеспечивать меры контроля, которые активно снижают риск загрязнения продукта.
Основная идея здесь состоит в том, чтобы понять продукт и процесс достаточно подробно, чтобы иметь возможность эффективно оценивать риски для качества продукта, связанные с загрязнением, и обеспечивать меры контроля, которые активно снижают риск загрязнения продукта.
Это достигается за счет ряда мер, начиная с эффективного проектирования, аттестации и валидации объекта и оборудования в сочетании с соответствующим количеством квалифицированного персонала с правильным отношением и опытом, чтобы иметь возможность эффективно управлять процессом. Во время производства продукта иметь системы для мониторинга и отслеживания производительности процесса, а также иметь системы для расследования отклонений процесса, которые могут повлиять на качество продукта, с использованием документированной системы оценки рисков. Чтобы организация могла действовать на основании данных оценки риска для исправления и предотвращения повторения отклонения, а квалифицированное лицо – иметь возможность эффективно принимать эффективные решения по отложению партии. После внедрения CCS его обслуживание станет частью периодической проверки качества продукции, чтобы подтвердить, что любые изменения в любой части процесса были внесены в соответствии с CCS и GMP. Также больше внимания уделяется оценке всех аспектов жизненного цикла продукта, включая поставку сырья и компонентов, а также транспортировку и хранение готовой продукции во время поставки на рынок.
Предпосылки
Этот раздел посвящен техническим аспектам проектирования и эксплуатации чистых помещений, включая использование барьерных технологий, перемещение людей / материалов в чистое помещение, очистку и дезинфекцию и демонстрацию поддержания подходящих условий окружающей среды во время аттестации чистого помещения.
Барьерные системы

В этот раздел внесено несколько существенных изменений, и первое из них заключается в том, что компании должны учитывать использование барьерной технологии (RABS или изоляторов) как части их CCS.
Мало кто будет спорить с тем, что барьерная технология обеспечивает более высокий уровень защиты продукта от микробного заражения при условии эффективного управления ими, однако существует множество различных конфигураций барьерных систем (открытые / закрытые системы), и в документе нет никаких указаний по выбору соответствующая система для защиты конкретного производимого продукта.
Для барьерных технологий необходимо учитывать попадание материалов / компонентов в критическую зону при сохранении окружающей среды класса А. CCS должен включать оценку процесса передачи материала и, если требуется, методов дезактивации. Также следует учитывать фоновую среду для RABS и изоляторов в зависимости от того, используются ли закрытые или открытые системы. В руководстве также указывается, что изоляторы отрицательного давления следует использовать только в том случае, если локализация продукта считается необходимой и применяются соответствующие меры контроля риска.
Требования к частоте проверки целостности барьерной системы и испытания перчаток на герметичность выполняются (минимум в начале и конце партии и после каждого вмешательства, которое может повлиять на целостность системы), а частота замены перчаток должна быть определена в CCS.
Введены требования к дезинфекции RABS и изоляторов (автоматизированные методы для изоляторов) и применению спороцидного агента для RABS (поскольку большинство из них являются открытыми системами), и должны быть доступны доказательства, подтверждающие, что дезинфицирующее средство не влияет на продукт.
Подача чистого воздуха
Есть несколько пунктов, связанных с подачей воздуха в чистое помещение, что оправдано, поскольку это одна из ключевых мер контроля по достижению требуемых условий окружающей среды для производства. К ним относятся обеспечение снабжения чистых помещений фильтрованным воздухом, поддержание положительного давления в соседних помещениях разного класса (минимум 10 паскалей) и эффективные системы мониторинга и предупреждения в случае отказа или снижения перепада давления между чистыми помещениями / изоляторами. В документе есть многочисленные ссылки на важность визуализации воздушного потока для проверки того, что воздушный поток внутри чистого помещения предотвращает проникновение частиц и действует для защиты продукта. Это требование, чтобы производились видеозаписи схем воздушного потока,
Квалификация оборудования для чистого и чистого воздуха
В заголовок этого раздела добавлено слово квалификация. Итак, чистые помещения GMP теперь необходимо квалифицировать и классифицировать, и между этими терминами проводится различие. Квалификация – это всеобъемлющий термин, описывающий полный набор тестов для демонстрации эффективного контроля окружающей среды, включая проверку целостности фильтра для мониторинга жизнеспособных и нежизнеспособных частиц, визуализацию воздушного потока, а также физических параметров, таких как мониторинг температуры и влажности. Классификация – это термин, применяемый к измерению мониторинга нежизнеспособных частиц. Что касается определения местоположений, сделана ссылка на серию стандартов ISO14644 для руководства в сочетании с оценкой критических местоположений в зоне A / B. В руководстве четко указано, что классификация нежизнеспособных частиц и микробиологическая оценка должны выполняться как в состоянии покоя, так и в рабочем состоянии. Предусмотрены пределы для микробиологического мониторинга во время аттестации, при этом основное изменение состоит в том, что для класса A требуется отсутствие роста на восстановительной среде (воздух, отстойные или контактные пластины), а не ранее указанная спецификация <1 КОЕ. Приведены минимальные требования к испытаниям для повторной аттестации, включая объем воздушного потока, подсчет нежизнеспособных данных, измерения давления воздуха и проверку целостности фильтров, поскольку тесты, определенные в 14664-2, являются информативными. Частота запланированных проверок для повторной аттестации определяется для областей уровня A / B и областей уровня C / D, а также приведены примеры, когда внеплановая повторная аттестация может быть оправдана, например, после серьезных изменений в чистом помещении.
Передача материалов и персонала
Больше внимания уделяется пониманию и контролю потенциальных путей заражения через объект с повышенными требованиями к перемещению материалов и персонала. К ним относятся более подробные сведения о необходимости разделения персонала и материалов, о перемещении персонала через зоны D в B и о необходимости мытья рук в раздевалке первой ступени. Усиленный контроль за перемещением материалов в зону класса A / B с новым требованием, согласно которому материалы должны быть в утвержденном списке, а передаточные люки должны быть защищены эффективной промывкой фильтрованным воздухом. По возможности, материалы, попадающие в асептические зоны, следует стерилизовать с помощью автоклавов или туннелей для депирогенизации или вводить с использованием эффективной системы дезинфекции с переносом, а газы или жидкости должны проходить через фильтр, задерживающий бактерии.
Дезинфекция
Название этого раздела было изменено с санитарии на дезинфекцию вместе с текущей отраслевой терминологией. Здесь есть новое требование, чтобы все процессы дезинфекции подтверждались исследованиями, демонстрирующими пригодность и эффективность дезинфицирующих средств в том виде, в каком они используются. Требование использования более чем одного типа дезинфицирующего средства сохраняется с уточнением использования периодического использования спорицидного средства. Сохраняются давние требования к стерильности моющих и дезинфицирующих средств при использовании в зоне A / B.
Оборудование
Этот раздел был изменен, чтобы уточнить необходимость иметь подробное описание конструкции оборудования во время валидации и поддерживать актуальную запись P&ID. По сути, компаниям необходимо обеспечить учет всех изменений в течение жизненного цикла оборудования. Что касается мониторинга производительности оборудования, методы должны быть определены на этапе URS, с отслеживанием тенденций в тревоге и системе во время работы. Процессы очистки должны быть валидированы.
Утилиты
В Приложении 1 2008 г. было только два пункта, касающихся коммунальных услуг. Последняя версия была значительно улучшена и включает особые требования для следующих
- Водные Системы
- Пар
- Газовые и вакуумные системы
- Системы отопления и охлаждения
В общем, оценка риска требуется для выявления предприятий с высоким риском (контакта с продуктом) с большим упором на мониторинг и поддержание тезисов. Текст о водных системах во втором проекте был изменен по сравнению с первым проектом с общим требованием, что производимая вода должна соответствовать текущей монографии соответствующей фармакопеи.

Имеются многочисленные ссылки на уменьшение образования биопленки в системе, например, на поддержание турбулентного потока. В соответствии с действующей европейской фармакопеей, WFI может производиться недистилляционными методами.
Требование стерилизации вентиляционных фильтров на резервуарах для воды, сделанное в первой редакции, остается, как и новое требование к периодической стерилизации или дезинфекции водной системы. Если слово стерилизация останется в тексте, это станет проблематичным, поскольку это будет невозможно продемонстрировать без использования биологических индикаторов, а необходимость в этом сомнительна. Существуют новые требования к микробиологическому мониторингу системы, включая необходимость определения мест наихудшего случая и необходимость отбора пробы в конце распределительного контура каждый день использования воды. Наконец, для систем WFI новое требование состоит в том, что водные системы должны включать системы непрерывного мониторинга, такие как TOC и проводимость, если это не обосновано.
Для пара, используемого в качестве прямого стерилизующего агента, качество воды, подаваемой в парогенератор, было уточнено как должным образом очищенное, а конденсат пара должен соответствовать действующей фармакопее для WFI. Физические параметры для испытания качества пара (неконденсирующийся газ, перегрев и доля сухости) должны периодически оцениваться. Газы, используемые в асептических процессах, должны быть отфильтрованы стерилизующим фильтром на месте использования.
Персонал
Этот раздел руководства был пересмотрен и расширен, чтобы включить требование о наличии достаточно квалифицированного, обученного персонала, имеющего опыт в производственных технологиях, для обеспечения соответствия GMP для стерильных продуктов. Существуют дополнительные требования к одежде и мониторингу, включая ежегодную начальную квалификацию персонала, а также оценку одежды и особые требования к обучению персонала чистых помещений в зависимости от критичности выполняемой работы.

В руководстве определены конкретные элементы обучения, которые включают:
- Основы микробиологии
- Гигиена
- Практика чистых помещений
- Методы контроля загрязнения
- Защита стерильных продуктов
- Исследования визуализации воздушного потока
Производство и особые технологии
В пересмотренный текст включен небольшой раздел о стерилизации терминалов, который практически не изменился по сравнению с текстом 2008 года. Значительно расширен раздел, посвященный асептической обработке.
Асептическая обработка и подготовка
Руководство требует, чтобы все аспекты асептического процесса были поняты и учтены в CCS, чтобы эффективно минимизировать риски загрязнения. Этому посвящен наш курс по управлению рисками микробного заражения в чистых помещениях, где риск заражения продукта количественно оценивается с помощью действующих мер контроля.
Следует рассмотреть использование технических мер для защиты продукта, таких как использование барьерных технологий и инженерных решений для уменьшения критических вмешательств и поддержания условий класса A во время передачи материалов / компонентов. Также существуют новые требования для определения подтвержденного времени выдержки стерилизованного оборудования, фильтрации продукта, времени наполнения и максимального времени, в течение которого открытые контейнеры находятся в критической зоне обработки.
Обработка стерильных продуктов
Существуют повышенные требования к герметизации и укупорке, включая поддержание условий окружающей среды и проверку целостности укупорочного средства, которые должны учитывать транспортировку и отгрузку. Для проверки продукта существуют новые требования к классификации дефектов, необходимость поддерживать библиотеку дефектов, которая фиксирует все известные классы дефектов, и потребность в результатах анализа тенденций.
Стерилизация
Некоторые давние требования к процессам стерилизации остались, например требование о ежегодной повторной валидации процессов тепловой стерилизации. Существуют дополнительные требования к выбору соответствующих биологических индикаторов для аттестации процессов стерилизации, а также повышенные требования, касающиеся подготовки и упаковки материалов для стерилизации. Для стерилизации компонентов многократного использования производителям необходимо учитывать поддержание условий класса A на этапах стерилизации путем соответствующего выбора обертки и переноса в зону класса A.
Для поверхностной паровой стерилизации компонентов существуют новые требования к оценке эффективности процесса во время валидации, включая расчет времени уравновешивания и корреляции между температурой и давлением. Кроме того, требования к проверке производительности системы, такие как проверка на герметичность (обычно еженедельно), когда цикл содержит вакуумную фазу, и метод обеспечения адекватного удаления воздуха (например, датчик воздуха). По завершении цикла следует оценить степень сухости загрузки путем визуального осмотра.
Требования к сухому теплу были усилены в связи с необходимостью понимания критических параметров процесса во время валидации. Сохраняется давнее требование продемонстрировать эффективность процесса депирогенизации во время валидации за счет снижения показателей эндотоксина на 3 логарифма.
Фильтрация
Для фильтрации продуктов, которые нельзя стерилизовать, необходимо полностью понимать этап фильтрации, поскольку это критическая контрольная точка в асептическом процессе. Стратегия фильтрации должна быть включена в CCS вместе с оценкой осуществимости избыточной стерильной фильтрации. Во время валидации этапа следует учитывать критические параметры или условия процесса, включая использование продукта и выбор наиболее подходящего бактериального заражающего организма во время тестирования удерживания.
Что касается проверки целостности фильтров, руководство требует проверки сборки стерилизованного фильтра перед использованием и после использования во время проверки целостности после завершения. Текст также предоставляет производителям возможность использовать оценку риска для обоснования методов, отличных от PUPSIT, для проверки целостности фильтра из-за ограничений процесса.
В этой ситуации производителей просят учитывать все риски загрязнения по всей цепочке поставок, методы проверки целостности и уровни бионагрузки продукта.
Заполнение формы и печать
Новые разделы, посвященные эксплуатации уплотнения с заполнением формы, требуют полного понимания критических параметров процесса, связанных с целостностью уплотнения во время валидации, и регулярного мониторинга прочности уплотнения.
Лиофилизация

Поскольку все большее количество производимых лекарственных препаратов в настоящее время представляют собой лиофилизированные биологические продукты, этой теме посвящен новый раздел. Руководство требует, чтобы все аспекты стадии лиофилизации учитывались в CCS, включая очистку, стерилизацию, загрузку разгрузочных флаконов и укупорку, чтобы гарантировать идентификацию опасностей с соответствующими средствами контроля. Опять же, здесь рассматриваются те же соображения, что и на других этапах, включая поддержание условий класса А во время переноса частично закрытых контейнеров, использование стерилизованных инструментов во время переноса и возможность нарушения схемы воздушного потока во время использования мобильных тележек.
Закрытые системы
В новом разделе, касающемся закрытых систем, представлены несколько элементов, которые следует учитывать в CCS, основная проблема заключается в том, чтобы система была разработана таким образом, чтобы гарантировать целостность. Производители также должны гарантировать, что система разработана таким образом, чтобы минимизировать количество ручных вмешательств и что система способна поддерживать стерильность. С точки зрения определения подходящих фоновых условий для закрытой системы, это снова сводится к оценке риска целостности системы и методу контроля целостности (например, испытание под давлением или мониторинг). Критичность процесса асептической / конечной стерилизации следует учитывать при определении соответствующей фоновой области для закрытой системы.
Системы одноразового использования
Системы одноразового использования все чаще используются в производстве, особенно для обработки биофармацевтических препаратов, из-за повышенной гибкости и уменьшения проблем, связанных с проверкой очистки. Возросшая зависимость от предварительно стерилизованных одноразовых установок и компонентов делает больший упор на управление поставщиками, поскольку обеспечение стерильности на месте было передано обратно поставщику. Таким образом, компании должны гарантировать, что, если этапы стерилизации проводятся третьей стороной, контроль и валидация будут соответствовать собственным политикам производителей в отношении стерилизации. Это ключевой элемент, который следует учитывать в стратегии CCS.
Жизнеспособный и нежизнеспособный мониторинг окружающей среды и процессов

Учитывая, что самый высокий процент недостатков GMP относится к системам качества, документации и расследованиям, неудивительно, что требования к этому были значительно усилены. Основное внимание в этом разделе уделяется необходимости полезного мониторинга, который дает значимые данные, которые можно отслеживать, анализировать и принимать меры для обеспечения непрерывного контроля производственной операции. Мониторинг окружающей среды может обеспечить только моментальный снимок производственных условий из-за неотъемлемых ограничений методов мониторинга, поэтому важно, чтобы были выбраны подходящие инструменты и подходящие среды, чтобы минимизировать неточности в достигнутых результатах.
Требуется, чтобы план экологического мониторинга определялся посредством оценки рисков. Соображения включают
- Места отбора проб (квалификация и регламент)
- Периодичность мониторинга
- Метод мониторинга (эффективен и не вносит загрязнения)
- Условия инкубации
- Исторические данные и существенные изменения
- Исследования визуализации воздуха
- Анализ тенденций, уровни предупреждений и действий
Одно из ключевых требований – действительно понимать данные и принимать соответствующие меры в случае увеличения числа нарушений уровня предупреждений / действий или последовательных нарушений или изменений микробной флоры на предприятии. Регулирующие органы четко заявили, что, когда данные мониторинга окружающей среды по событиям отзыва впоследствии были проанализированы после события, часто наблюдается значительный сдвиг в микробном профиле в пределах категорий D и C, который, если он обнаружен в то время, мог предотвратить загрязнение продукта. событие происходит.
Нежизнеспособный мониторинг
Сохраняется необходимость контролировать 5-микронные частицы в зоне класса А. Дальнейшие пояснения представлены в отношении непрерывного мониторинга среды класса A с требованием контролировать продолжительность критической обработки, включая настройку. В руководстве поясняется, что объем выборки не обязательно должен быть таким же, как для классификации, а объем выборки требует обоснования. Наконец, пересмотренный текст включает в себя необходимость проверки всех данных, полученных в результате первоначальной квалификации и текущего мониторинга, для проверки соответствующих уровней предупреждений и действий при эксплуатации.
Мониторинг жизнеспособных частиц (мониторинг микробов)
Неудивительно, что уровень мониторинга сводится к риску загрязнения на этапе процесса с повышенным уровнем мониторинга в областях класса A / B в асептических процессах. Целью плана микробного мониторинга является определение подходящих участков в производственных операциях, которые предоставляют значимые данные, чтобы гарантировать, что соответствующие условия окружающей среды поддерживаются. Места мониторинга должны быть близки к месту проведения работ, поэтому требуется анализ операций в производственных зонах для выявления зон с интенсивным движением, горячих точек, потенциальных мертвых зон, критических контактных поверхностей, зон с высокой проходимостью и поверхностей в чистом помещении. CCS должен учитывать все потенциальные источники загрязнения и маршруты передачи по всему объекту, чтобы сосредоточить внимание на местах мониторинга на наиболее важных операциях. Были добавлены повышенные требования к мониторингу персонала с учетом мониторинга персонала после критических вмешательств и при выходе из зоны класса B (перчатки, халаты и рукава). Для микробиологического мониторинга воздуха эффективность методов отбора проб требует квалификации. Раздел также включает руководство по применению быстрых или автоматизированных методов после демонстрации эквивалентности или превосходства установленной методологии (традиционные методы, основанные на культуре).
Предел действия для обнаружения микробов в зонах класса A был изменен на «отсутствие роста», и было дано разъяснение, что рост любых микробов в результате мониторинга в зоне класса A требует расследования. Также дается разъяснение относительно необходимости идентифицировать микробы, восстановленные в областях класса A и B, до уровня вида, и что следует учитывать идентификацию микробов, обнаруженных в областях C и D, вне тренда или вне спецификации (OOS ) События.
Моделирование асептического процесса (APS или наполнение средой)
Текст 2008 года содержал 5 пунктов, посвященных теме наполнения носителями, по меньшей мере кратких для такого важного аспекта асептической обработки. Этот раздел теперь был расширен до 19 пунктов с требованиями о том, как проводить следы, асептические манипуляции, вмешательства и действия, которые необходимо предпринять в случае отказа медиа-следа.
С точки зрения проведения наполнения средой требуется план, чтобы определить задачи, которые необходимо выполнить, а также необходимость имитации фактического процесса как можно точнее, включая такие факторы, как переключение смен. Требование к минимальному количеству контейнеров, которое должно быть оценено посредством оценки риска, чтобы гарантировать, что все действия и вмешательства включены, типичное минимальное количество 5 000 и 10 000 единиц (бывшее для малых партий) остается неизменным. Дальнейшие рекомендации представлены по инкубации заполненных единиц и идентификации микробов, выделенных из зараженных единиц. Из руководства очень ясно, что цель во время заполнения среды – нулевой рост. В случае восстановления загрязненного агрегата требуется расследование, которое определяет наиболее вероятную первопричину, соответствующие корректирующие меры, план повторения следа и, конечно, оценка всех связанных записей с момента последнего успешного медиа-заполнения.
Контроль качества (QC)
Этот раздел начинается с требования о необходимости в должным образом обученном персонале с опытом работы в микробиологии во время проектирования и эксплуатации стерильного производственного процесса. Во многих ситуациях требуются подготовленные микробиологи для проведения исследований и оценки микробиологических данных, однако полезно, чтобы весь персонал, занимающийся производством стерильных продуктов, имел базовую подготовку по микробиологии. Это позволит инженерам оценить конструкцию помещения / оборудования или не микробиологов, а также активно участвовать в исследованиях или иметь мнение о микробиологических данных.
Любая оценка требуется для всего сырья, компонентов и продуктов, для которых риск микробного заражения был идентифицирован в CCS. Существуют новые требования к периодическому мониторингу бионагрузки для процессов окончательной стерилизации, даже если были приняты методы избыточного уничтожения. Для окончательно стерилизованного продукта добавлено новое требование о том, чтобы пробы для испытаний на стерильность отбирались из мест наихудшего случая. Наконец, требуются письменные планы относительно действий, которые необходимо предпринять, когда результаты мониторинга окружающей среды выходят за рамки тенденции или отсутствуют.
Резюме и заключение
Для любого, кто принимал участие в обзоре Приложения 1, очевидно, что это переписывание, а не просто пересмотр. Процесс консультаций все еще продолжается, и окончательный документ не был опубликован, однако проекты консультаций дают очень хорошее представление о том, что будет включать окончательный документ. На вопрос, являются ли предложения более ясными для понимания или менее двусмысленными? Ответ во многом сводится к интерпретации и точке зрения читателя. Текст 2008 года содержал 127 пунктов, и при их изучении часто было очень мало деталей или разъяснений в поддержку требований. Пересмотренный текст содержит около 300 статей с гораздо более высоким уровнем детализации и написан как дорожная карта элементов, которые следует учитывать в CCS для производителей стерильных продуктов и регулирующих органов.